
Research Article
The first survey of distribution of inherited deafness patterns in individuals referred to genetic center of Ahvaz welfare organization, Southern Iran
Ali Mohammad Foroughmand1, Hamid Galehdari1, Gholamreza Mohammadian2, Abdolrahman Rasekh3, Jasem Ghavabesh2
1- Department of Genetics, Faculty of Sciences, Shahid Chamran University of Ahvaz, Iran
2- Welfare Organization of Ahvaz, Genetic Counseling Centre, Iran
3- Department of Statistics, Faculty of Mathematical Sciences and Computer, Shahid Chamran University of Ahvaz, Iran
Received: 10 January 2011, accepted: 1 June 2011
Abstract
Background and Aim: Deafness is a heterogeneous disorder induced by genetic and environmental factors. It is the most common hereditary sensory-neural disorder that affects 1/1000 to 1/2000 of the newborns. More than 70% of hearing loss cases are caused by genetic disorders, 85% of which result from nonsyndromic autosomal recessive sensory-neural hearing loss. Up to now, more than 100 genes contributing in hearing loss have been determined. Alteration of these genes may result in hearing loss.
This study was performed to identify the inheritance patterns of deafness and its relation with ethnicity, gender and consanguineous marriages.
Methods: In this survey, data from 356 families affected by hearing loss and referred to welfare organization of Ahvaz during the time were collected based on sex, ethnic groups and relativeness.
Results: The results state a high frequency of autosomal recessive deafness caused by consanguineous marriages within Arab and non-Arab ethnic groups (p<0.05). But no significant difference in gender.
Conclusion: In conclusion, the high frequency of autosomal recessive deafness among the population with a high frequency of consanguineous marriages is considerable. The dominant pattern of deafness observed in this population was autosomal recessive.
Keywords: Syndromic deafness, non-syndromic deafness, inheritance patterns, consanguineous marriages, Arab, non-Arab population
مقاله پژوهشي
بررسي توزيع الگوهاي کمشنوايي ارثي در کمشنوايان مراجعهكننده به مرکز مشاورۀ ژنتيك سازمان بهزيستي اهواز
عليمحمد فروغمند1، حميد گلهداري1، غلامرضا محمديان2، عبدالرحمن راسخ3، جاسم غوابش2
1ـ گروه ژنتيك، دانشكده علوم، دانشگاه شهيد چمران اهواز، ایران
2ـ مرکز مشاوره ژنتيك، بهزيستي اهواز، ایران
3ـ گروه آمار، دانشكده علوم رياضي و کامپيوتر، دانشگاه شهيد چمران اهواز، ایران
چكيده
زمينه و هدف: در بيش از 70 درصد، کمشنواییها به دليل ناهنجاريهاي ژنتيكي ايجاد ميشود. تاكنون بيش از 100 مكان ژني گزارش شده است که تغيير آنها ميتواند به کمشنوايي منجر گردد. هدف از اين بررسي، تعيين فراواني الگوهاي توارثي در جمعيت کمشنواي مراجعهکننده به سازمان بهزيستي اهواز و ارتباط آن با عوامل ديگر از قبيل قوميت، جنسيت و ازدواجهاي خویشاوندی بود.
روش بررسي: در اين بررسي پرونده 356 خانواده مراجعهكننده به مرکز مشاوره ژنتیک سازمان بهزيستي اهواز مورد مطالعه قرار گرفت. افراد مبتلا به کمشنوايي با توجه به قوميت، جنسيت و همچنين رابطۀ خويشاوندي والدين آنها با هم در دستجات مختلف و براساس الگوي توارثي حاصل از بررسي نحوۀ انتقال کمشنوايي در شجرۀ خانوادههاي کمشنوا، تقسيمبندي گرديد.
يافتهها: توارث اتوزومي نهفته؛ فراوانی بیش از 60 درصد، رايجترين الگوي ژنتيکي بود. فراوانی اغلب انواع ازدواجهای خویشاوندی در جمعیت غیرعرب بیشتر از عرب بود(05/0p<) تفاوت معنیداری بین دو جنس دیده نشد(05/0p>).
نتيجهگيري: درکنار مشاهده تفاوتهاي بين دو جمعيت در فراواني ازدواجهاي خويشاوندي تفاوت معنيداري بين کمشنواييهاي توارثي و کمشنواييهاي حاصل از عوامل محيطي وجود داشت و الگوي غالب کمشنوايي در منطقه الگوي آتوزومي نهفته است.
واژگان كليدي: کمشنوايي سندرمي، کمشنوايي غيرسندرمي، الگوي توارثی ازدواج خويشاوندي، عرب، غيرعرب
(دریافت مقاله: 20/10/89، پذیرش: 11/3/90)
مقدمه
كاهش شنوايي و کمشنوايي از شايعترين نقصهاي حسي در انسان است كه از هر1000 تا 2000 نوزاد يك مورد را تحت تأثير قرار ميدهد(5-1). يكي از برنامههاي اصلي سازمان جهاني بهداشت (WHO) تشويق كشورها براي ارائه و اجرای برنامههايي در قالب طرح ملي پيشگيري از اختلال شنوایی و نقص شنوايي است. در همين رابطه تلاش برنامههاي بينالمللي بر این است که اختلالات جدي شنوايي در سنین پایینتر تشخیص داده شود(6و7).
بر اساس بر آوردي از Prasad و همکاران(8) و Storm و همکاران(9) تقريباً نيمي از کمشنواييهاي دوران كودكي منشأ ژنتيكي دارند، و همچنین بر اساس گزارش Willems 90 درصد موارد اين نقص به دليل اختلالات ژنتيكي ايجاد ميشوند(10).
بيش از 10 سال است كه نقش اختلالات ژنتيكي در بروز اختلالات شنوايي شناسايي شده است، و تاكنون صدها ژن در اين ارتباط گزارش شده است(1و13-11). پايۀ ارزيابي ژنتيكي، تشخيص دقيق حالتهاي مشكوك است. ارزيابي ژنتيكي براي
كودكان کمشنوا كه به تازگي تشخيص داده شدهاند بايد مورد توجه باشد، بهویژه اگر هيچ علت خاصی تعيين نشده باشد. بهعنوان مثال، خانوادۀ كودكي كه به كاهش شنوايي متعاقب مننژيت مبتلا شده است به ارزيابي ژنتيكي نياز ندارد(11).
اختلالات شنوايي ژنتيكي به دو دستۀ کمشنوايي ژنتيكي سندرمي و غيرسندرمي تقسيم ميشوند. تاكنون بيش از 400 سندرم شناسايي شده است كه در آنها اختلال شنوايي به دنبال ابتلا به بيماري و همراه ساير علائم بهوجود میآید(11و14). اما در دستۀ دوم، یعنی کمشنواييهاي ژنتيكي غيرسندرمي، درصد بالايي از موارد کمشنوايي بدون ظهور ناهنجاريهاي ديگر بروز ميكند، و معمولاً يك ژن منفرد در بروز اين عارضه نقش دارد و در آن الگوهاي متفاوت وراثتی کروموزومي و ميتوکندريايي مشاهده ميشود(11).
جمعيت خوزستان تركيبي از قوميتهاي مختلف از جمله فارس و عرب است. اين تركيب جمعيتي ميتواند بر توزيع صفات ژنتيكي و بيماريهاي ارثي تأثيرگذار باشد. شناخت ويژگيهاي ژني جمعيتي و بيماريهاي رايج در ميان اين جمعيتها ميتواند در توسعۀ راههاي آموزش و جلوگيري از گسترش اختلالات ژنتيكي اهميت ويژهاي داشته باشد. نگرش كلي به انواع کمشنواييها در اهواز با توجه به الگوي وراثتي و همچنين جمعيت عرب و غيرعرب در همين راستا ميتواند مفيد و قابل توجه باشد. اين بررسي ميتواند به سؤالاتي پيرامون الگوي وراثتي کمشنواييهاي شايع و شناخت قوميتي كه ابتلای بيشتري دارد پاسخ دهد. هدف از اين بررسي تعيين فراواني الگوهاي وراثتي در جمعيت کمشنواي مراجعهکننده به مرکز مشاورۀ ژنتیک سازمان بهزيستي اهواز در سالهاي 1382-1374 و ارتباط آن با عوامل ديگر از قبيل قوميت، جنسيت و ازدواجهاي خویشاوندی است.
روش بررسي
در اين بررسي پروندۀ 356 خانواده كه به دليل دارا بودن افراد کمشنوا (در دامنۀ بین dB 90-56) در خانواده و یا خويشاوندان براي مشاورۀ ازدواج يا بارداري در سالهاي 1382 تا 1374 به بهزيستي اهواز مراجعه کرده بودند مورد مطالعه قرار گرفت. نیمی از این جمعیت را خانوادههای عرب تشکیل میدادند. اثبات کمشنوا بودن افراد مبتلا قبلا توسط متخصصان و با استفاده از دستگاههاي تخصصي انجام گرفته بود (دستگاه ادیومتر مدل AC5 شرکت Interacoustic، کشور دانمارک و دستگاه ادیومتر ایمیتانس مدل AZ26شرکت Madsen کشور دانمارک). با توجه به اطلاعات فردي 650 تا 657 فرد کمشنوا در زمینۀ قوميت (عرب و غیرعرب)، جنسيت و همچنين رابطۀ خويشاوندي والدين
آنها با هم در دستجات مختلف تقسيمبندي شدند. در اين بررسي با توجه به الگوي توارث كه از طريق رسم شجرۀ خانوادهها صورت گرفت، سعي شد تا افراد مبتلا بر اساس رابطۀ خويشاوندي، نوع الگوي وراثتی، موارد محيطي و تكگير و جنسيت افراد طبقهبندي شوند. برای بررسيهاي آماري از آزمون کاي دو استفاده گرديد.
يافتهها
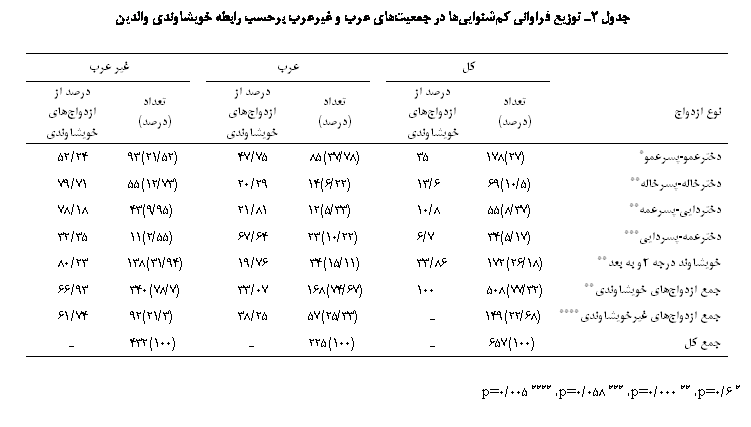
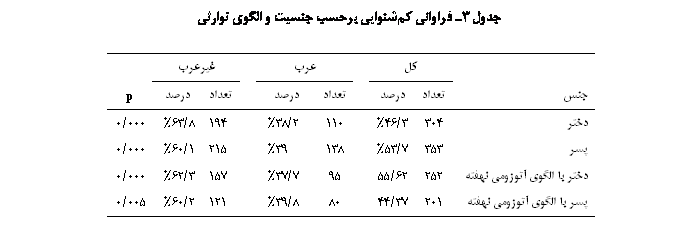
در اين بررسي بيش از 60 درصد کمشنوايان استان خوزستان داراي الگوي وراثتی اتوزومي نهفته بودند (جدول 1). اين مقدار بيش از مجموع تعداد ديگر الگوهاي توارثي آتوزومي بارز و وابسته به جنس نهفته بود (001/0p<). از طرف ديگر، موارد کمشنوايي ناشی از عوامل محيطي و تكگير در مقايسه با الگوي آتوزومي نهفته فقط حدود 14 درصد را در برميگرفت (جدول 1)، اگرچه این دو الگو تفاوتهای معنیداری بین دو جمعیت مورد بررسی نشان میدهند. نكته ديگر در اين رابطه، فراواني مبتلايان غيرعرب در مقايسه با افراد عرب است. ازدواجهاي خویشاوندی با فراواني بيش از 77 درصد در جمعيت هدف مشاهده گرديد (جدول 2). همچنين در اين جدول فراواني خويشاوندي براي هر نوع ازدواج (دخترعموـپسرعمو و بقيۀ انواع ازدواجها) و براي مجموع ازدواجها محاسبه و مقايسه شد که به جز برای دخترعمو و پسرعمو برای بقیۀ موارد ازدواجهای خویشاوندی تفاوتهای معنیداری مشاهده شد (جدول 2). در عين حال ازدواجهاي خويشاوندي درجۀ 1 با بيش از 50 درصد و ازدواجهاي خويشاوندي درجۀ 2 و به بعد در غيرعرب بيشتر و در مجموع فراواني ازدواجهاي خويشاوندي در غيرعرب 61 درصد بود. از طرف ديگر، توزيع جنس کمشنوايي در هر دو جنس دختر و پسر تقريباً نزديك به هم بود، اگر چه تفاوتهای قابل توجهی بین دو جمعیت از نظر فراوانی جنسی قابل مشاهده است (جدول 3). جدول 4 نشان ميدهد که تفاوت قابل توجهي بين فراواني کمشنواييهاي ارثي و کمشنواييهاي حاصل از عوامل محيطي وجود دارد.
بحث
کمشنواييهاي ژنتيكي بهصورت آتوزومي بارز و نهفته و وابسته به X نهفته و بارز و همچنين با الگوي ميتوكندريايي مشاهده ميشوند، كه با توجه به گزارشهای قبلي شايعترين فرم، كاهش شنوايي غيرسندرمي حسيـعصبي با الگوي آتوزومي مغلوب است(15و16). اگرچه الگوهاي وراثتي و موارد تكگير و محيطي افراد غيرعرب توزيعي همانند الگوي افراد عرب را نشان ميدهند. به عبارت ديگر، الگوي وراثت با قوميت بستگي ندارد، ولي فراواني افراد مبتلا در افراد غيرعرب قابل توجه است.
 در مجموع، با توجه به
فراواني الگوي آتوزومي نهفته در بين هر دو جمعيت و همچنين الگوي توارث وابسته به
جنس نهفته ضرورت مشاورۀ ژنتيك در اين جمعيتها برای جلوگيري از گسترش
عارضۀ کمشنوايي امري ضروري و غيرقابل اغماض است. فراواني كم افراد کمشنوا
در دستههاي تكگير و محيطي در اين بررسي احتمالاً به دليل عدم ارجاع اين افراد به
بهزيستي و بخش ژنتيك است. برای ريشهيابي اين تفاوت انجام مطالعات جدي لازم
است. ولي بهصورت ابتدايي تفاوتهاي كمّي جمعيتي، ارجاع دادن افراد به مراكز مشاورۀ
ژنتيك، توجه به اهميت مشاورۀ ژنتيك در قوميتهاي متفاوت ميتواند از جمله
عوامل مؤثر باشد.
در مجموع، با توجه به
فراواني الگوي آتوزومي نهفته در بين هر دو جمعيت و همچنين الگوي توارث وابسته به
جنس نهفته ضرورت مشاورۀ ژنتيك در اين جمعيتها برای جلوگيري از گسترش
عارضۀ کمشنوايي امري ضروري و غيرقابل اغماض است. فراواني كم افراد کمشنوا
در دستههاي تكگير و محيطي در اين بررسي احتمالاً به دليل عدم ارجاع اين افراد به
بهزيستي و بخش ژنتيك است. برای ريشهيابي اين تفاوت انجام مطالعات جدي لازم
است. ولي بهصورت ابتدايي تفاوتهاي كمّي جمعيتي، ارجاع دادن افراد به مراكز مشاورۀ
ژنتيك، توجه به اهميت مشاورۀ ژنتيك در قوميتهاي متفاوت ميتواند از جمله
عوامل مؤثر باشد.
همانطور كه در جدول 1 مشاهده میشود فراواني کمشنواييهاي با الگوي وراثت آتوزومي نهفته بيشترين كمّيت را به خود اختصاص ميدهد. اين توزيع الگوي وراثتی ميتواند از نوع ازدواجهاي خويشاوندي در منطقه تأثير پذيرد. در همين راستا مطالعات در بعضي از كشورها توسط محققانی مانند Tabchi و همکاران (2000) در عربستان و Fageeh(2003) در لبنان مؤید همين مطلب است(17و18). رجوع به جدول 2 و مشاهدۀ ازدواجهاي خویشاوندی با فراواني بيش از 77 درصد بدون شك ميتواند دليلي قوي براي اين استدلال باشد. نكتۀ جالب ديگر در اين رابطه، فراواني ازدواجهاي خويشاوندي درجۀ 1 با بيش از 50 درصد است. در اين ازدواجها بيشترين فراواني اختصاص به ازدواجهاي دخترعموـپسرعمو دارد كه بيش از 50 درصد اين ازدواجها در ازدواجهاي درجۀ 1 و 35 درصد ازدواجهاي خویشاوندی به اين گروه تعلق دارد.
با مقايسۀ نوع ازدواجهاي خويشاوندي در والدين افراد کمشنوا در جمعيتهاي عرب و غيرعرب مشاهده ميشود كه ازدواجهاي خويشاوندي دخترخالهـپسرخاله، دخترداييـپسرعمه، خویشاوند درجۀ 2 و به بعد در غيرعرب خيلي بيشتر و ازدواج دخترعموـپسرعمو تقريباً برابر و دخترعمهـپسردايي در قوميت عرب بيشتر است، و در مجموع فراواني ازدواجهاي خويشاوندي در غيرعرب با حدود 67 درصد رقم قابل توجهي است. وجود اين الگوها علاوه بر توجه بيشتر متخصصان مربوط، توجه بيشتر مشاوران ژنتيك و دستگاههاي اطلاعرساني سلامتي و متخصصان ژنتيك مولكولي و پزشكي را ميطلبد.
توزيع جنسيتی کمشنوايي در هر دو جنس دختر و پسر تقريباً نزديك به هم است. عدم وابستگي الگوي توارثي به جنسيت از ويژگيهاي الگوهاي وراثت آتوزومي نهفته است، و همان طور که انتظار ميرود تفاوت جنسيتي در اين نتايج مشاهده نميشود، اگرچه تعداد دخترها و پسرهاي مبتلا در جمعيت غيرعرب به دلايلي که روشن نيست داراي تفاوت معنيداري با جمعيت عرب است (جدول 3).
نکته ديگري که در اينجا و مطالعات بعدي بايد مورد توجه قرار گيرد، تفاوت قابل توجه بين کمشنواييهاي ارثي و کمشنواييهاي حاصل از عوامل محيطي است. این تفاوت هم بین الگوها و هم برای الگوهای کمشنوایی بین دو جمعیت مورد مطالعه کاملا مشهود است (جدول 4). در مجموع، جداول فوق با نشان دادن نسبت الگوهاي وراثتی و شيوع قابل توجه الگوي آتوزومي نهفته ميتوانند در ترسيم يك خطمشي كلي براي مطالعات مولكولي مرتبط با ژنهاي درگير در کمشنواييها تعيينكننده باشند. از طرف ديگر، اهميت توجه مشاوران ژنتيك به الگوي غالب کمشنوايي در منطقه بين دو جمعيت عرب و غيرعرب را نشان ميدهد. همچنين ميتواند براي خانوادههاي داراي سابقۀ خویشاوندی زنگ خطري باشد تا بيشتر به مشاورههاي قبل از ازدواج اهمیت دهند(11و19). يادآوري خطرات احتمالي همراه ازدواجهاي خويشاوندي و زمينهسازي براي كاهش اين ازدواجها، و كنترل تعداد اولاد خانوادههاي در خطر از طريق ارتقای فرهنگ عمومي در دستيابي به اين اهداف ميتواند نقش قابل توجهي داشته باشد.
نتیجهگیری
در مجموع ميتوان گفت که درکنار مشاهدۀ تفاوتهاي بين دو جمعيت در فراواني ازدواجهاي خويشاوندي تفاوت معنيداري بين کمشنواييهاي ارثي و کمشنواييهاي حاصل از عوامل محيطي وجود دارد، و الگوي غالب کمشنوايي در منطقه الگوي آتوزومي نهفته است. البته با ارتقای فرهنگ در زمینۀ ازدواجهای خویشاوندی و توسعۀ مشاورۀ ژنتیک در سطح کشور، بهخصوص برای خانوادههای در معرض خطر، میتوان به کنترل و کاهش اختلالات ارثی از جمله کمشنوایی امیدوار بود.
REFERENCES
1. Modamio-Høybjør S, Moreno-Pelayo MA, Mencía A,·del Castillo I, Chardenoux S,·Armenta D, et al. A novel locus for autosomal dominant nonsyndromic hearing loss (DFNA44) maps to chromosome 3q28-29. Hum Genet. 2003;112(1):24-8.
2. Nance WE. The genetics of deafness. Ment Retard Dev Disabil Res Rev. 2003;9(2):109-19.
3. Falk MM. Biosynthesis and structural composition of gap junction intercellular membrane channels. Eur J Cell Biol. 2000;79(8):564-74.
4. Marazita ML, Ploughman LM, Rawlings B, Remington E, Arnos KS, Nance WE. Genetic epidemiological studies of early-onset deafness in the U.S. school-age population. Am J Med Genet. 1993;46(5):486-91.
5. Cohen MM, Gorlin RJ. Epidemiology, etiology, and genetic patterns. In: Gorlin RJ, Toriello HV, Cohen MM, editors. Hereditary Hearing Loss and its Syndromes. NY: Oxford University Press; 1995. p. 9-21.
6. Kudo T, Ikeda K, Oshima T, Kure S, Tammasaeng M, Prasansuk S, et al. GJB2 (connexin 26) mutations and childhood deafness in Thailand. Otol Neurotol. 2001;22(6):858-61.
7. Nekahm D, Weichbold V, Welzl-Mueller K, Hirst-Stadlmann A. Improvement in early detection of congenital hearing impairment due to universal newborn hearing screening. Int J Pediatr Otorhinolaryngol. 2001;59(1):23-8.
8. Prasad S, Cucci RA, Green GE, Smith RJ. Genetic testing for hereditary hearing loss: connexin 26 (GJB2) allele variants and two novel deafness-causing mutations (R32C and 645-648delTAGA). Hum Mutat. 2000;16(6):502-8.
9. Storm K, Willocx S, Flothmann K, Van Camp G. Determination of the carrier frequency of the common GJB2 (connexin-26) 35delG mutation in the Belgian population using an easy and reliable screening method. Hum Mutat. 1999;14(3):263-6.
10. Willems PJ. Genetic causes of hearing loss. N Engl J Med. 2000;342(15):1101-9.
11. Robson CD. Congenital hearing impairment. Pediatr Radiol. 2006;36(4):309-24.
12. Murgia A, Orzan E, Polli R, Martella M, Vinanzi C, Leonardi E, et al. Cx26 deafness: mutation analysis and clinical variability. J Med Genet. 1999;36(11):829-32.
13. Harris KC, Erbe CB, Firszt JB, Flanary VA, Wackym PA. A novel connexin 26 compound heterozygous mutation results in deafness. Laryngoscope. 2002;112(7 Pt 1):1159-62.
14. Sasanfar R, Tolouei A, Hoseinipour A, Farhud DD, Dolati M, Hoghooghi Rad L, et al. Frequency of a very rare 35delG mutation in two ethnic groups of Iranian populations. Iranian J Publ Health. 2004;33(4):26-30.
15. Reardon W. Genetic of deafness. J Med Genet. 1992;29(8):521-6.
16. Lefebvre PP, Van De Water TR. Connexins, hearing and deafness: clinical aspects of mutations in the connexin 26 gene. Brain Res Brain Res Rev. 2000;32(1):159-62.
17. Tabchi B, Rassi B, Akl E, Fares G. Epidemiology of profound neurosensory deafness in Lebanese children. J Med Liban. 2000;48(5):294-7. French.
18. Fageeh NA. Prospective study of hearing loss in schools for deaf children in Assir region, Saudi Arabia. West Afr J Med. 2003;22(4):321-3.
19. Erbe CB, Harris KC, Runge-Samuelson CL, Flanary VA, Wackym PA. Connexin 26 and connexin 30 mutations in children with non-syndromic hearing loss. Laryngoscope. 2004;114(4):607-11.